Baskerville is always looking to showcase and to advertise the great work that is being produced on Baskerville.
If you are a Baskerville user and would like your work displayed here please contact
baskerville-tier2-support@contact.bham.ac.uk
Author: Kit Windows-Yule, Associate Professor in Chemical Engineering, University of Birmingham
What is this project about?
As a Turing Fellow, I have had the good fortune to lead or otherwise be involved in a number of Data Study Groups (DSGs). DSGs are a unique sprint research activity run by the Alan Turing Institute, in which a group of ~10 PhD-level data science and AI experts from diverse fields come together to address a significant challenge proposed by an industrial or governmental “Challenge Owner”. In one of the more notable projects, the Challenge Owner was Rolls Royce, and the aim of the challenge was to use AI methods to try and develop new ideas for ways to optimise the production of turbine blades, minimising the waste and cost associated with defective parts.
In order to operate optimally, a whole turbine blade must be formed from a single crystal – that is, all its constituent atoms must be organised neatly into a single crystalline lattice. Without such single crystal components, which are capable of operating in the very hottest parts of an aeroplane engine, it is impossible to manufacture the modern jet engine. Despite decades of advances, challenges remain to identify and overcome reasons for failure to achieve a single crystal in each and every casting. Because of the actions of multiple mechanisms that result in formation and growth of secondary grains, the experiments required to separate them are too costly and too slow to perform in practice. As such, a data-driven approach to solving this problem could prove transformative.
During the DSG, a variety of artificial intelligence and data science methods were tested, including convolutional neural networks (CNNs), generative adversarial networks (GANs), and topological data analysis. While the complete solution to such a complex problem could not realistically be obtained within the DSG timeframe, several proposed solutions showed significant promise and, as such, funding was acquired to continue exploring these promising methods in a PhD project stemming from this initial exploratory work.
Why was Baskerville chosen?
For two main reasons – the scale of the challenge, and the sensitivity of the data.
Concerning the former, the data provided by Rolls Royce included over 3GB of process data and around 185GB of 3D images. In order to process these data and use them in the development and training of novel AI models within the restrictive one-week timeframe of the DSG, significant compute power was required. Baskerville’s 208 NVIDIA A100 GPUs provided the ideal solution to this issue.
Regarding the latter, despite being desensitised, the data used for the study represented real Rolls Royce process data, and as such were decidedly commercially sensitive. The secure environment provided by Baskerville allowed the data to be handled and processed safely, without fear of data leaks. Without the security promises offered by Baskerville, this project could not have gone ahead.
How has Baskerville been useful?
In addition to the need for extreme compute power and data security mentioned above, the system’s flexibility and ease of use were also pivotal to the project’s success. Due to the nature of the DSG, researchers from different institutes around the world had to learn how to use an HPC that they had never worked on before within a very limited timeframe. The extensive documentation and training resources available to the participants, as well as the excellent support provided by the Advanced Research Computing (ARC) team, ensured that they could get up to speed in no time at all, and thus make full use of the time and resources available to them.
Author: Matthew Foulkes, Professor of Physics, Imperial College London
What is your research about?
I am a theoretical materials physicist, interested in all types of everyday
matter, from molecules to metals, insulators, semiconductors, magnets, and even
superconductors. My research ranges from practical and applied at one end -- I
study the formation and properties of protective oxide scales on
high-performance alloys containing aluminium -- to theoretical and abstract at
the other end -- I recently worked on superconductivity in an idealised model
system known as the unitary Fermi gas. The common theme is that most of my
research projects start from the quantum mechanical many-particle Schrödinger
equation, which serves, in effect, as a “grand unified theory" for materials
physics. There are more fundamental layers (quantum field theories and string
theories) below it, but for the purposes of a materials physicist the
many-particle Schrödinger equation is all we need.
Gaining the ability to solve the Schrödinger equation accurately and
efficiently would be transformative, allowing materials scientists, physicists,
chemists and biochemists to participate in the simulation-based revolution that
has already overtaken much of engineering. This would greatly reduce the cost
of discovering and developing new materials, chemicals, catalysts, and
medicines. Perhaps unsurprisingly, the many-particle Schrödinger equation is
difficult to solve accurately for any but the smallest systems. In practice, we
almost always rely on mean-field-like approximate methods such as density
functional theory. Most of the limitations in our understanding of molecules
and materials, such as our failure to explain high-temperature
superconductivity in the cuprates, arise when these approaches fail.
Four years ago, working with a small team from Google DeepMind, we introduced
FermiNet [1], a new neural-network-based approach to solving the many-particle
Schrödinger equation. Similar approaches had been tried for model systems, but
ours was the first successful application to real molecules and solids. The
FermiNet neural network takes any set of particle positions as input and
returns the corresponding value of the many-particle wavefunction. In practice,
the network replaces the analytic trial wavefunctions used in many other
computational approaches to quantum theory. The weights and biases that define
the network are optimised using the variational principle, which states that
the best attainable approximation to the ground-state wave function can be
found by minimising the energy expectation value. No external data are
required. The computational cost is high, but FermiNet and its successor the
Psiformer [2] are much more accurate than density functional theory, scale
better with system size than comparably accurate quantum chemistry approaches,
and run very naturally on massively parallel GPU-based supercomputers such as
Baskerville.
Recent work, much of which used Baskerville, has shown that neural wave
functions are able to describe strongly correlated systems beyond the purview
of DFT, and that they can “discover” quantum phase transitions, such as Wigner
crystallisation and the onset of superconductivity in the electron gas, without
human guidance [3,4]. They seem to be particularly good at describing unusual
systems, where conventional methods struggle. Our recent work on the binding of
positrons to molecules, for example, produced what we believe to be the most
accurate computations of binding energies and annihilation rates available [5].
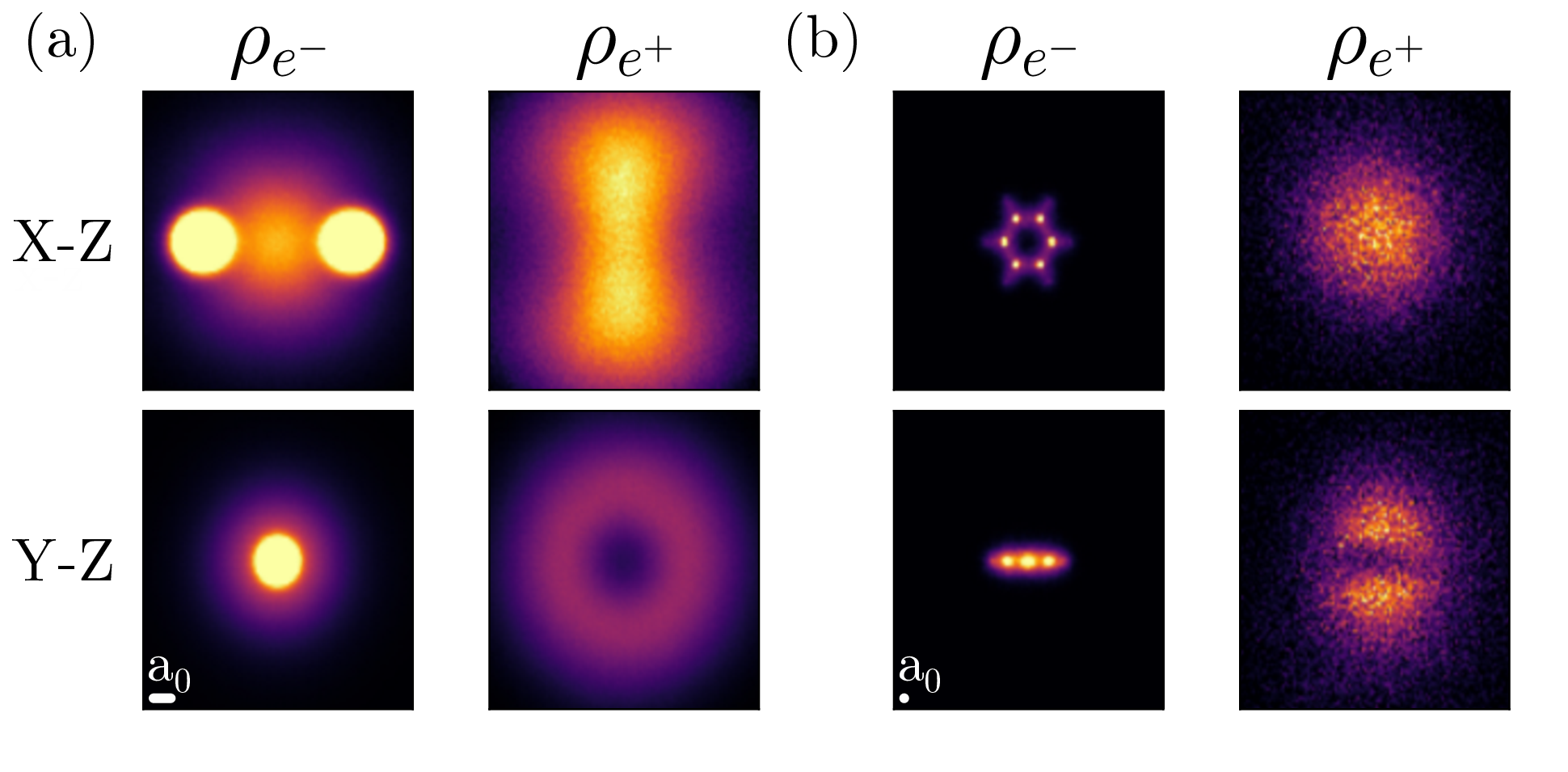
Positrion Desity of Li2 Benzene
The ground-state electron and positron densities of positronic (a)
dilithium and (b) benzene molecules. The left-hand/right-hand columns show the
electron/positron density. In the dilithium molecule, the positron density
resembles a doughnut wrapping the covalent bond. In the benzene molecule, the
positron density is very diffuse, resembling a bun surrounding the benzene
hamburger. The scale bar at the bottom left of each subfigure corresponds to
the radius of a hydrogen atom.
Why did you choose Baskervile?
Our codes, developed in close collaboration with researchers at Google
DeepMind, were designed to run on GPU supercomputers from the start, leveraging
their vast low-level parallelism via the JAX library (roughly equivalent to
NumPy with the addition of automatic differentiation and just-in-time
compilation), the XLA accelerated linear algebra machine learning compiler, and
NVIDIA's CUDA programming model. Our simulations are memory hungry, with large
runs requiring 40GB A100 GPUs or better. The GPU resources available to
university-based researchers in the UK are limited, and were even more limited
when we applied for our first Baskerville grant via the EPSRC's Access to High
Performance Computing mechanism. Baskerville was one of only two A100 machines
available at the time, and the other was already full up.
Although we had no choice but to apply for time on Baskerville, we were happy
to discover that it is powerful, stable and well run. The support we received
was always helpful and efficient and the installation of a few large-memory
80GB A100 GPUs was a boon. Much of our research over the past few years would
have been impossible without Baskerville. If only it were 10 times larger!
D. Pfau, J.S. Spencer, A.G.D.G. Matthews, and W.M.C. Foulkes, Phys. Rev. Research 2, 033429 (2020).
I. von Glehn, J.S. Spencer, and D. Pfau, https://arxiv.org/abs/2211.13672v2 (2022).
G. Cassella, H. Sutterud, S. Azadi, N.D. Drummond, D. Pfau, J.S. Spencer, and W.M.C. Foulkes, Phys. Rev. Lett. 130, 036401 (2023). (Editors’ Suggestion).
W.T. Lou, H. Sutterud, G. Cassella, W.M.C. Foulkes, J. Knolle, D. Pfau, and J.S. Spencer, Phys. Rev. X 14, 021030 (2024).
G. Cassella, W.M.C. Foulkes, D. Pfau and J.S. Spencer, Nat. Commun. 15, 5214 (2024).